Sequence databases and similarity search
This repository contains the materials for the “Databases in ecology and comparative genomics course”: day 1. It deals with the primary Nucleotide (and protein) databases and the way to interrogate them!
Github URL: https://github.com/claudioametrano/sequence_databases_similarity_search
Software required (on HPC)
BLAST 2.16.0
NCBI datasets
HMMER3.4
Trimal
Iqtree2
mafft
Software required (locally)
A fasta file reader (Aliview, MEGA, Jalview, Bioedit …)
Terminal (Linux and Mac), Mobaxterm (Win), to ssh into the HPC and GUI file client (e.g. Filezilla, Mobaxterm)
iTOL (works from browser)
BEFORE WE START
Login to your account on the HPC and start an interactive session (as we won’t run long analyses that require to submit a job to the cluster queue):
ssh username@l2.gsc1.uni-graz.at
srun --mem=8G --ntasks=1 --cpus-per-task=4 --time=10:00:00 --pty bash
download this repository:
git clone https://github.com/claudioametrano/sequence_databases_similarity_search
Rename the folder containing the results, so you won’t overwrite it running the analyses of this tutorial, and it will be available if needed:
cd sequence_databases_similarity_search
mv results results_backup
mkdir results
We will work on the HPC cluster (High Performance Computing) using Singularity and containers which have the specific software we need (often from Biocontainers.pro; Biocontainers paper)
You can either run commands interactively with the container staying open (singularity shell):
Download the container
$ singularity pull https://container_address/container_name
Run container interactively
$ singularity shell container_name # Start the shell showing Sungularity> as long as you stay in the container
$ commands you want to run
$ exit # to exit the container
… or just execute a single command (singularity exec) in the container, useful if you need to run something for longer
$ singularity exec container_name command_to_run
It is also possible to install the needed software via Conda, in this case the commands can be run without singularity run or singularity exec, but you will need to install Anaconda/Miniconda and the following software (maybe in a dedicated Conda environemnt) before starting:
conda install conda-forge::mamba
mamba install bioconda::blast=2.16.0
mamba install conda-forge::ncbi-datasets-cli=18.2.0
mamba install bioconda::hmmer=3.4
mamba install bioconda::trimal=1.5.0
mamba install bioconda::iqtree=2.4.0
mamba install bioconda::mafft=7.525
1- Nucleotide Sequence Databases
Sequence databases are scientifically invaluable, they serve as comprehensive repositories for DNA (and proteins) submitted by researchers. They provide centralized, standardized, and easily searchable collections of genetic data (from genes to entire genomes), enabling organism identification (DNA barcoding and meta-barcoding), evolutionary studies, comparative and functional genomics. As important as the database, is the way we query them to obtain results suitable for our analyses.

Let’s have a look at the available resources! National Center for Biotechnology Information: NCBI
What are the different databases?
| Database | Description |
|---|---|
| PubMed | Biomedical and life-science citations and abstracts. |
| PubMed Central / PMC | Full-text open-access biomedical and life-science articles. |
| Bookshelf | Books, reports, manuals, GeneReviews, and NCBI documentation. |
| MeSH | Controlled vocabulary used to index PubMed articles. |
| NLM Catalog | Records for journals, books, and other NLM collection items. |
| Nucleotide | DNA and RNA sequences from GenBank, RefSeq, TPA, PDB-linked records, and other sources. |
| RefSeq | Curated, non-redundant reference sequences for genomes, transcripts, and proteins. |
| Genome | Genome-level information organized by organism and sequencing project. |
| Assembly | Genome assembly records |
| SRA — Sequence Read Archive | Raw high-throughput sequencing reads from genomics, transcriptomics, metagenomics, and amplicon studies. |
| BioProject | Project-level metadata linking related sequencing studies, samples, reads, and assemblies. |
| BioSample | Sample-level metadata such as organism, isolate, tissue, environment, location, and experimental source. |
| Taxonomy | Organism names, taxonomic classification, synonyms, and NCBI TaxIDs. |
| Gene | Gene-centered information including loci, transcripts, proteins, homologs, phenotypes, and publications. |
| GEO — Gene Expression Omnibus | Functional genomics data such as RNA-seq, microarray, expression, and molecular abundance datasets. |
| GEO DataSets | Curated datasets derived from GEO submissions. |
| GEO Profiles | Individual gene expression or molecular abundance profiles from GEO data. |
| Protein | Protein sequences from RefSeq, GenPept, Swiss-Prot, PIR, PRF, PDB, and other sources. |
| CDD — Conserved Domain Database | Conserved protein domains, domain models, alignments, and functional annotations. |
| Identical Protein Groups | Groups of identical protein sequences across organisms and databases. |
| Protein Family Models | Protein family models, including HMMs and conserved architecture-based classifications. |
| Structure / MMDB | Experimentally determined 3D biomolecular structures. |
| dbSNP | Short genetic variants such as SNPs, small indels, and microsatellites. |
| dbVar | Large structural variants such as CNVs, deletions, insertions, inversions, and translocations. |
| ClinVar | Human genetic variants and their reported clinical significance. |
| dbGaP | Genotype–phenotype association studies |
| MedGen | Medical genetics concepts linked to genes, variants, diseases, and publications. |
| GTR — Genetic Testing Registry | Information about genetic tests and testing laboratories. |
| OMIM | Human genes and genetic disorders, integrated with NCBI resources. |
| PubChem Compound | Chemical structures and compound-level records. |
| PubChem Substance | Depositor-submitted chemical substances and descriptions. |
| PubChem BioAssay | Biological screening assays and activity results for chemical substances. |
| PubChem Pathways | Molecular pathways linking genes, proteins, chemicals, and biological processes. |
In the main page search for an organism you would like to know more about and check the results.
TASK 1
Let’s now create some queries in the nucleotide database (Select Nucleotide, then click search then “advanced”):
Progressively refine your query with the following steps:
Create a simple query that search for all the DNA sequences belonging to truffles
Only for sequence of ribosomal RNA
Limit the length range to exclude sequences much longer than rRNA
Exclude environmental sequences
Visualize one sequence in GenBank format, and in fasta
Download all of them as a fasta file ordered by sequence length and rename it to “tuber_rDNA.fas” (upload in in “results/task1/” it will come handy later)
Hints: You can use logical “AND”, “OR” and “NOT”; To search a combination of more than one word consecutively put them in “()”; Most of the filters are also available on the left column of the search page
Questions:
What is the number of sequences retrieved for each step with the query becoming more stringent?
Are these sequences representing the same locus/i? How would you assess this? Why they vary so much in length?
2- Sequence Alignment
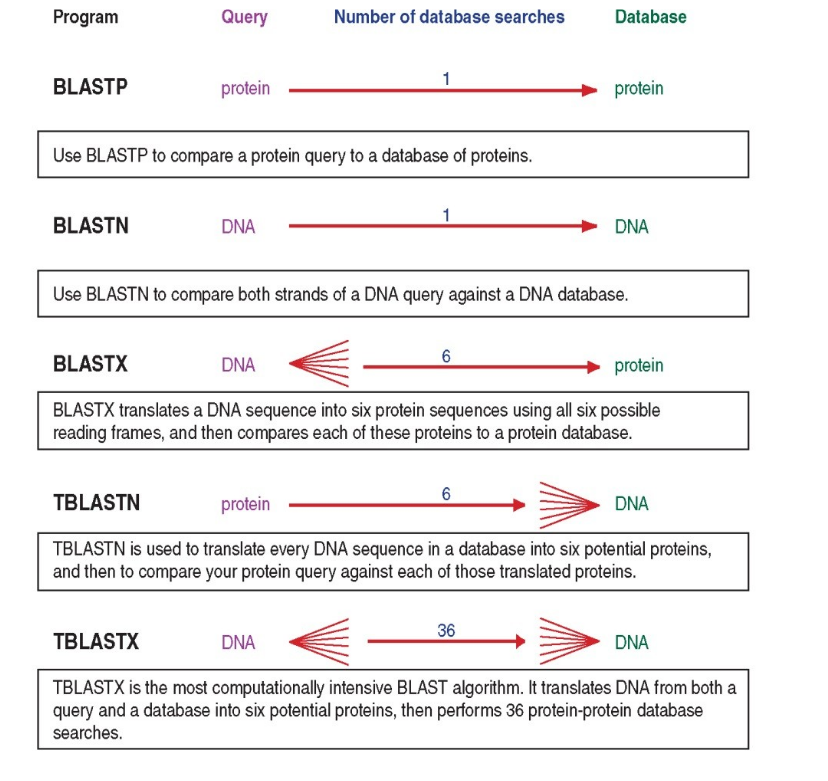
Now, let’s say you just got your shiny new sequence from an unknown organism out of a Sanger sequencing run, most of the time, we want to interrogate these database with a query that is a sequence itself, not a species name. Then, we need alignment algorithms that aligns our query sequence with those in the database. One widely used example is Basic Local Alignment Search Tool: BLAST It has the enormous advantage of being integrated in NBCI as web-service. Let’s take a look at what it can do!
 credit: allan.haldane@temple.edu
credit: allan.haldane@temple.edu
The BLAST algorithm and scoring system
Highly similar sequences will have stretches of similarity, we can pre-screen the sequences for common long stretches. This idea is used in BLAST by breaking up the query sequence into W-mers.
BLAST algorithm steps:
Split query into overlapping words of length W (the W-mers, same as kmers)
Find exact matches: the seeds ->Lookup each word in indexed table to find the location in the database where the seeds occur.
Extend the seeds (Smith-Waterman algorithm for local alignment) until the score of the alignment drops off below some threshold (local to global alignment).
Report matches with overall highest scores -> what score?
Bit Score: Quality of alignment, higher score -> better alignment; Normalized measure of similarity, BLAST first computes a raw alignment score S from the substitution matrix + gap penalties you chose. That raw score is then put on a universal “bits” scale so different scoring systems can be compared:
$$ S’ = \frac{\lambda S - \ln K}{\ln 2} $$
E-Value : Expected value measures the expected number of “random” sequences with a score S or better in a random sequence database of equal size. Lower E-value = less likely the hit was a false-positive -> it might have a biological relationship with the query sequence (but can only infer homology with this, not prove it).
$$ E = K \cdot m \cdot n \cdot e^{-\lambda S} $$
Where:
m is the length of the query sequence.
n is the length of the database (i.e., the sum of all the lengths of all the sequences in the database).
K and λ normalize the alignment score for the type of scoring matrix and gap penalties.
S is the alignment score. It is calculated based on the selected scoring matrix and the given sequence alignment. The score reflects the sum of substitution and gap scores for the aligned residues.
Expectation value (E-value): Containing m and n, E value also normalize for the length of the query and of the database.
BLAST algorithm parameters

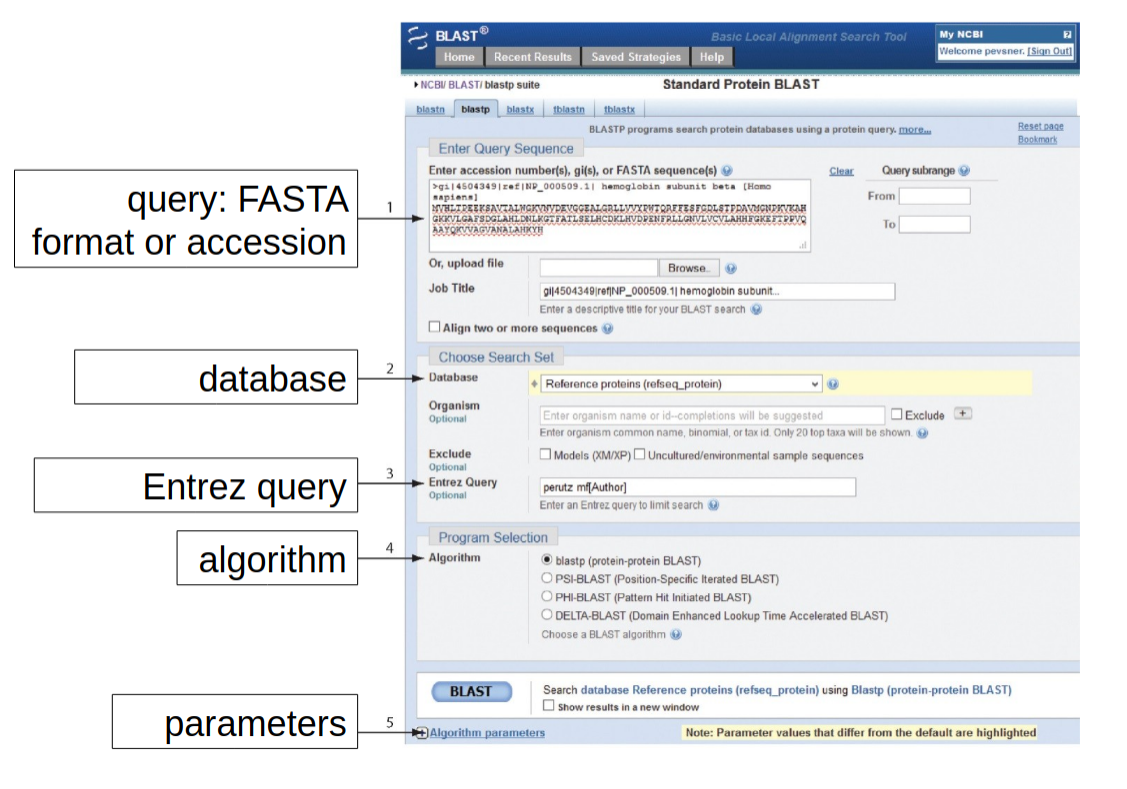
2a- Use of BLAST+ suite via web browser
TASK 2
Open the blast_me.fasta file and look what kind of sequence it is (DNA or AA), then select the most suitable BLAST service
Load the /data/blast_me.fasta file (or copy/paste the content from less or cat )
Start the BLAST alignment with everything left at default
Check results, what is it?
Now try to find it by BLAST in the following species: mouse, rat, chimpanzee, humans, chicken, zebrafish, mosquito, fruit fly and baker’s yeast (hints: use scientific name, relax blast parameter if needed!)
Download a fasta for each (check query cover!) and put them in results/task2 togheter with your blast_me.fasta
Optional: Build a phylogenetic tree with this tiny dataset
Put everything in a multi-fasta file
Align using mafft (container at https://depot.galaxyproject.org/singularity/mafft:7.525–h031d066_1)
Revise your alignment by eye before and after deleting not well aligned columns (use Trimal, container at https://depot.galaxyproject.org/singularity/trimal:1.5)
Use iqtree2 to infer the Maximum Likelihood tree (container at https://depot.galaxyproject.org/singularity/iqtree:2.3.6–h503566f_1)
Visualize you tree with iTOL. iTOL:iteractiveTreeOfLife. You probably already have a rough idea on how related are these organism, but in case you do not iTOL has a nice Tree of Life welcoming you on their website, compare it with the one you got!
Does your tree reflect the known phylogenetic relationships among those organisms? If not, can you think why this may happen? How would you improve this phylogeny?
2b- Use of BLAST+ Command Line Interface (CLI)
manual here
Why should I learn command line BLAST when BLAST via browser is so convenient?
BLAST on custom database (e.g. something new you sequenced)
Automation: run it on server/cluster, as part of a bioinformatic automatized workflow
Speed and limit of online BLAST (e.g. number of max sequences per submission (200), maximum allowed length (1Mb) )
We will learn how to use the command line blast tools, also known as BLAST+. We will use it to:
Blast search NCBI databases
Build our own blast database
Retrieve information from blast hits
Local BLAST
$ singularity pull https://depot.galaxyproject.org/singularity/blast:2.16.0--h66d330f_4
Show available pre-packaged databases available on NCBI servers by the helper script
$ singularity exec blast:2.16.0--h66d330f_4 update_blastdb.pl --showall
To download a db from NCBI:
$ mkdir results/NCBI_databases
$ singularity exec blast:2.16.0--h66d330f_4 update_blastdb.pl SSU_eukaryote_rRNA
$ mv *.gz *.md5 ./results/NCBI_databases/
$ cd results/NCBI_databases
$ md5sum -c *.md5 # check downloaded files integrity
$ cd -
Download some compact databases for common rDNA barcode markers (update_blastdb.pl can take a space-separated list): SSU_eukaryote_rRNA LSU_eukaryote_rRNA ITS_eukaryote_sequences Check for errors in downloads and if the number of files checks out (sometimes NCBI server does not respond on time)
Example of protein-protein BLAST (blastp) query example:
singularity exec blast:2.16.0--h66d330f_4 blastp -query query.fasta -db path/to/db_name -out blastout_query_vs_db_name.txt
TASK 3
BLAST using the appropriate blast tool the unknown sequence contained in data/sequence.fasta against each of the BLAST databases in /data/NCBI_BLAST_databases:
Questions:
Which kind of sequence is it? How long is it?
With what blast command would you start? Is any other blast tool suitable?
Why you are not getting meaningful results? How can you try solve it? (hint: did you run it at default parameters? check the -help for the use of: -reward -penalty -gapopen -gapextend -word_size; or check the -task argument )
What locus/i is it? How can you visualize this? (hint: do you have “by chance”, among the file we produced, one that can help?)
To what organism does the sequence belong?… Can you tell without any doubt?
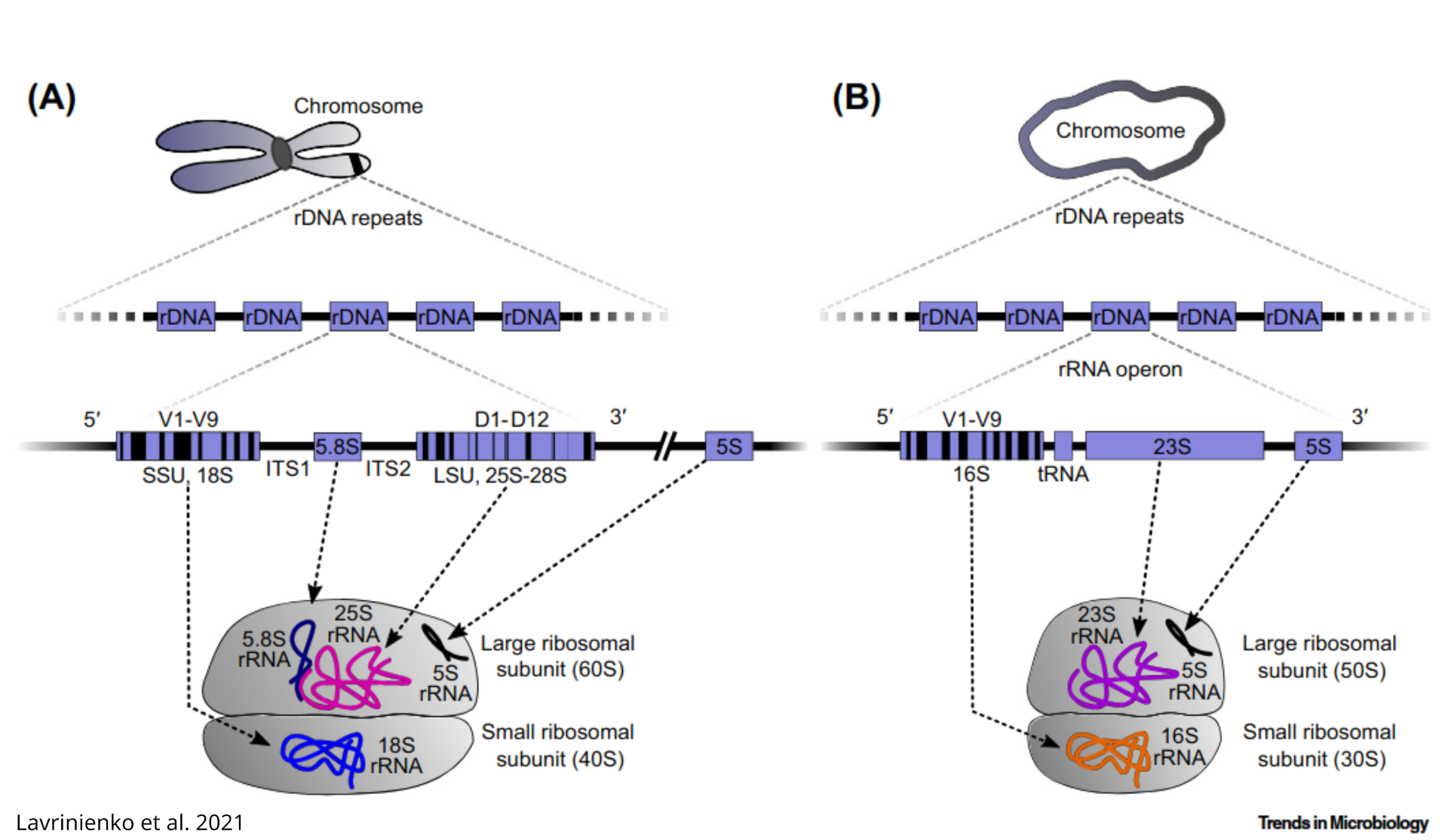
ADD ON: build a MSA (Multiple sequence alignment)
After discovering which locus was contained in the query sequence, to clearly delimit which ITS we can build a MSA that more clearly shows the boundaries of each locus in rDNA, for example using task1 tuber_rDNA.fas file:
# get the first (the longest, if you sorted them in the beginning) seqs from file
$ head -n 1000 ./results/task1/tuber_rDNA.fas > results/task3/tuber_rDNA_longest_plus_sequence.fas
# add the query sequence
$ cat ./data/sequence.fasta >> results/task3/tuber_rDNA_longest_plus_sequence.fas
# Here you can grep -c the fasta, to see how many you got
$ singularity exec mafft\:7.525--h031d066_1 mafft --thread 4 results/task3/tuber_rDNA_longest_plus_sequence.fas > results/task3/tuber_rDNA_longest_plus_sequence_mafft_aligned.fas
download and visualize in aliview (or similar software). The file in results_backup is manually edited to keep only fewer meaningful sequences, as downloaded sequence are often non-overlapping portions of rDNA. It is an ITS1!
Remote blast in command line!
As some BLAST databases are huge (hundreds of Gbs to some Tb) you can still BLAST remotely (exactly as you would do on NCBI website using a browser), let’s try a BLASTn using the complete nucleotide Genbank collection
$ singularity exec blast:2.16.0--h66d330f_4 blastn -remote -query ./data/sequence.fasta -db nt -out ./results/task3/blastout_sequence_vs_nt.txt
$ less ./results/task3/blastout_sequence_vs_nt.txt
How the results differ from what you got so far? Why? If you check the databases you used locally, searching for the Tuber species from you sequence you will find out…
$ singularity exec blast:2.16.0--h66d330f_4 blastdbcmd -db ./results/NCBI_databases/ITS_eukaryote_sequences -entry all | grep "Tuber "
$ singularity exec blast:2.16.0--h66d330f_4 blastdbcmd -db ./results/NCBI_databases/ITS_eukaryote_sequences -entry all | grep "Tuber melanosporum"
NOTE
Your results are dependent both on the method and the reference database!
Bring Your Own Database (BYO~~B~~D)
Building you custom database can be useful in several occasions, in general, every time the search you need to perform using BLAST is not contained in one of the default NCBI databases
Let’s create a BLAST database using as input the genome assembly of Tuber melanosporum downloaded from NCBI Genome/dataset under accession GCA_000151645.1 , but it can also be a genome you just assembled or whatever other collection of sequences you want to perform similarity searches on.
From command line (works as well from the NCBI webpage under genome/dataset)
$ mkdir results/task4
$ wget -P results/task4 https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/151/645/GCF_000151645.1_ASM15164v1/GCF_000151645.1_ASM15164v1_genomic.fna.gz
unzip and build the database
# exteact using gzip
$ gunzip results/task4/GCF_000151645.1_ASM15164v1_genomic.fna.gz
# extract using 7zip
$ 7z x ./results/GCF_000151645.1_ASM15164v1_genomic.fna.gz -o./results/^C 7z x ./data/ncbi_dataset.zip -o./results/
# Build the database from the assembly fasta
$ singularity exec blast:2.16.0--h66d330f_4 makeblastdb -in ./results/task4/GCF_000151645.1_ASM15164v1_genomic.fna -dbtype nucl -out ./results/task4/GCF_000151645.1_assembly_db
now try to query the newly created database with ./data/sequence.fasta, what do you expect?
$ singularity exec blast:2.16.0--h66d330f_4 blastn -query ./data/sequence.fasta -db ./results/task4/GCF_000151645.1_assembly_db -out ./results/task4/blastout_sequence_vs_GCA_000151645.1.txt
Extract BLAST results from the database
A more efficient way to store (and re-use in bioinformatic workflows) your blast results, and extract aligned sequences
$ singularity exec blast:2.16.0--h66d330f_4 blastn -query ./data/sequence.fasta -db ./results/task4/GCF_000151645.1_assembly_db -out ./results/task4/blastout_sequence_vs_GCF_000151645.1_tabfmt.txt -outfmt "6 qseqid sseqid qstart qend sstart send sstrand evalue bitscore pident qcovs sseq"
$ cat results/task4/blastout_sequence_vs_GCF_000151645.1_tabfmt.txt
-outfmt 6 has its own default fields, but you can customize it, as we just did listing the information we want in the output file
TASK 4
Parse the previous blast result:
How many copies of rDNA are present in the assembly?
Can you tell if they are located on the same chromosome using this assembly?
redirect the hit in a fasta file
awk -F'\t' '!seen[$2]++ {print ">" $2 "_ITS1" "\n" $NF}' ./results/task4/blastout_sequence_vs_GCF_000151645.1_tabfmt.txt > ./results/task4/sequence_vs_GCF_000151645.1_first_hit.fasta
!seen[$2]++ only keeps the first result per subject name (“$2” is the second tab separated field), $NF selected the last field (NF: number of fields), in this case the sequence.
3- The right method for every occasion: other commonly used alignment/mapping tools
BLAST is not the only tool available, some examples of widely used alignment/mapping tools are summarized below. Alignment/mapping, and in general search by sequences similarity is one of the most frequent activity in bioinformatics, so literally a plethora of algorithms exist and are developed.
| Use-case | Tool | main feature |
|---|---|---|
| Rapid protein (large databases, metagenomics) | DIAMOND; |
faster than BLAST (in fast mode, various speed/sensitivity setting) |
| Amplicon clustering/search, small-to-medium nucleotide search | USEARCH | faster than BLAST with similar recall |
| Short-read DNA mapping (Illumina 50–300 bp) | BWA; Bowtie2 |
fast mapping of millions of reads on reference sequence, output in SAM/BAM |
| Long-read / assembly alignment (ONT, PacBio, ≥ 1 kb reads) | Minimap2 | mix of speed, accuracy, and splice awareness |
| Remote homology search (detecting distant protein families/motifs) | HMMER3 | sensitivity for weakly conserved domains |
| Spliced transcript / protein to genome alignment | EXONERATE | Good at finding intron/exon boundaries |
3a HMMER
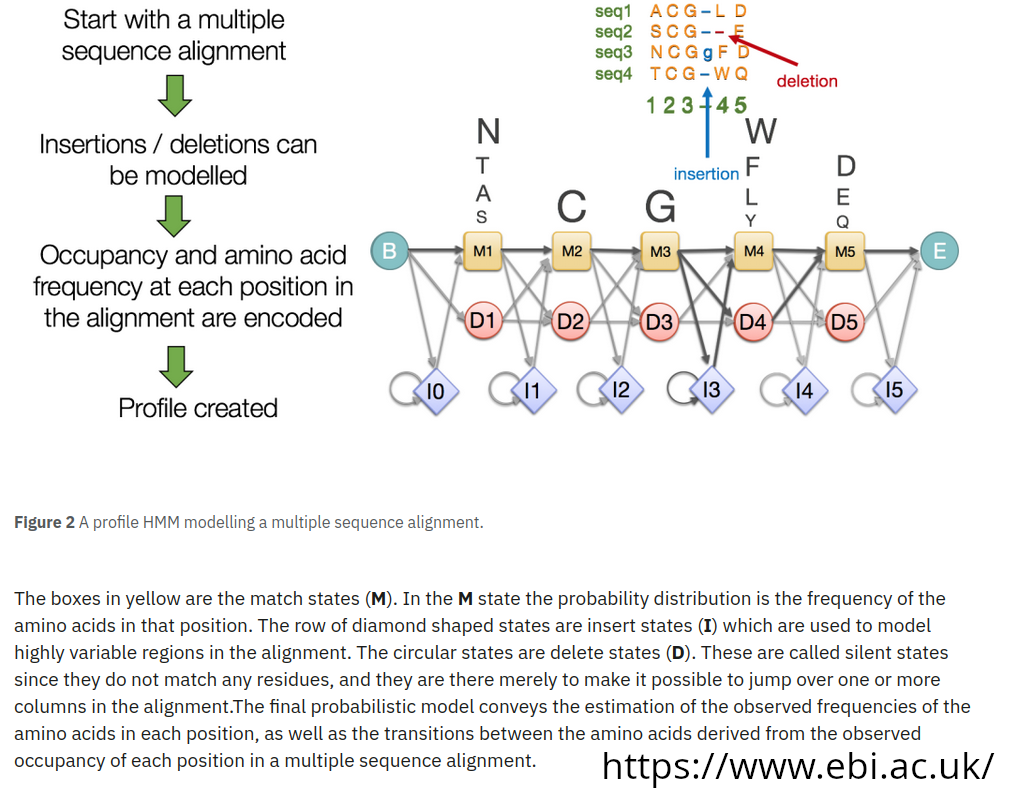
Used to search sequence databases for (possible) homologs of protein sequence. Its aim is to sum up all the information and columns variability of a multiple sequence alignment, modelling substitution probabilities, insertion and deletion.
It is based on probabilistic models known as profile hidden Markov models (profile HMMs) –> it can use a profile based on a multiple-sequence alignment (MSA) to captures which positions are conserved or variable across a gene. A profile turns those position-specific patterns into quantitative scores.
Profile hidden Markov models (profile HMMs) are statistical models of the primary structure consensus of a sequence family. They can model both insertions and deletions, they cope better than a single sequence with “gappy” sequence evolution. WHAT IS HIDDEN, THEN?: the state path that generated the sequence we observe.
It needs a trusted MSA (as less biased as possible toward specific taxa) to build an effective profile that can obtain homologous sequences from a wide range of organisms.
HMMs do have limitations. For example they cannot capture any higher-order correlations. An HMM assumes that the identity of a particular position is independent of the identity of all other positions (e.g. does not include scoring terms for nearby amino acids in a three-dimensional protein structure)
Difference to BLAST and other pairwise alignment algorithms
Profile HMMs are statistical descriptions of the consensus of a multiple sequence alignment. They use position-specific scores for amino acids (or nucleotides) and position specific scores for opening and extending an insertion or deletion. Traditional pairwise alignment (for example, BLAST (Altschul et al., 1990), FASTA (Pearson and Lipman, 1988), or the Smith/Waterman algorithm (Smith and Waterman, 1981)) uses position-independent scoring parameters. This property of profiles captures important information about the degree of conservation at various positions in the multiple alignment, and the varying degree to which gaps and insertions are permitted.
The seven states modeled (page 210 ofHMMER manual for accurate description of .hmm file format): m: match d: deletion i: insertion
Possible transitions: m->m, m->i, m->d, i->m, i->i, d->m, d->d …
what is missing? d->i and i->d are not modeled (it simplify the model without reducing its power)
Example of users of HMMER profiles:
Open an HMM profile and check yourself (from BUSCOs OrthoDB genes sets)
$ wget -P results/ https://busco-data.ezlab.org/v5/data/lineages/ascomycota_odb12.2025-07-01.tar.gz
# extract only one profile from the BUSCO set
$ tar -xvzf results/ascomycota_odb12.2025-07-01.tar.gz -C results/ --wildcards '*685555at4890.*'
Two file format, one position-specific residue frequencies (from AUGUSTUS)
less -S ./results/ascomycota_odb12/prfl/685555at4890.prfl
…and the actual profile-HMM file used for BUSCO search
less -S ./results/ascomycota_odb12/hmms/685555at4890.hmm
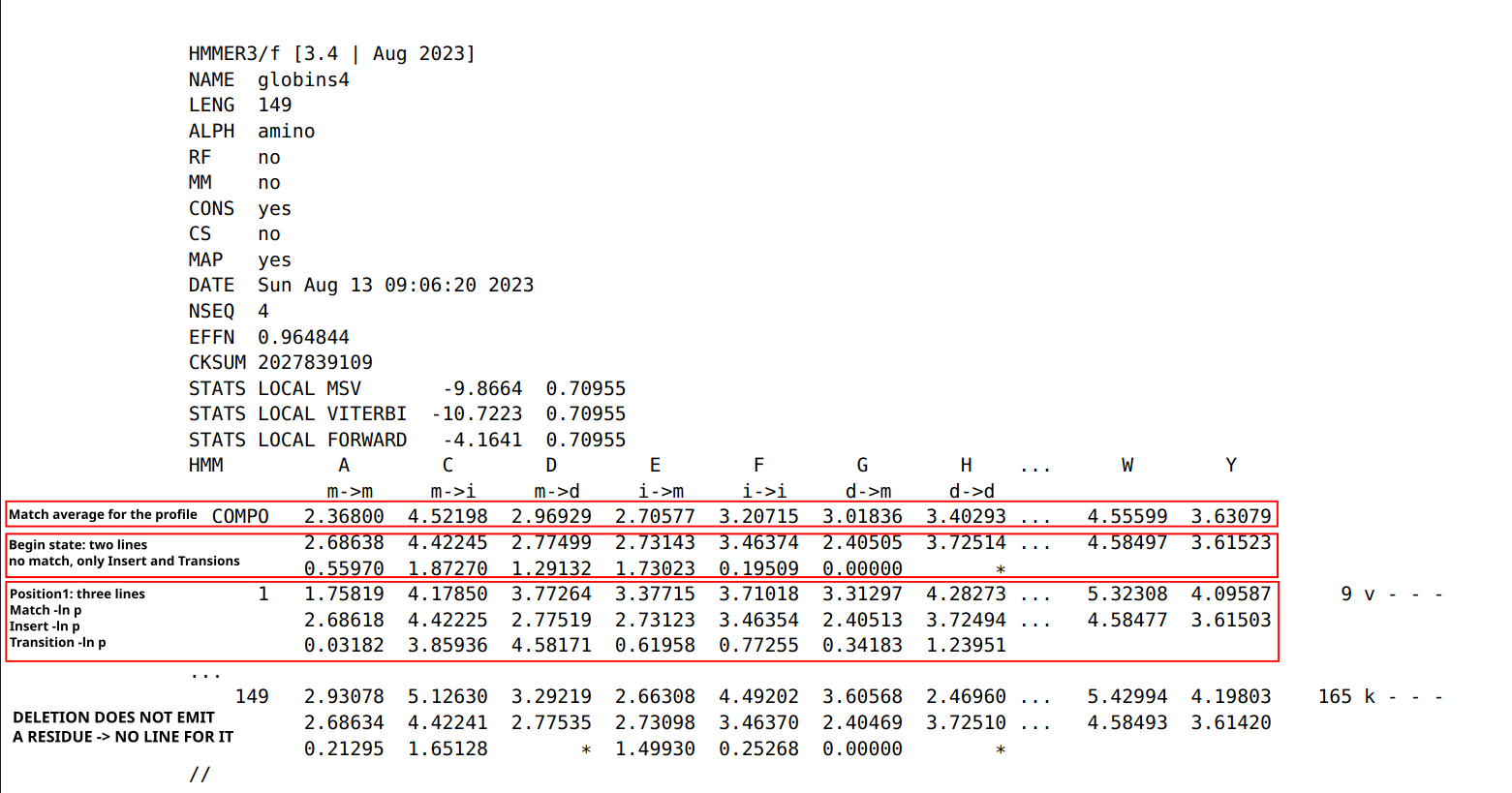
Building a profile:
select amino-acid sequences from a gene of interest(in /data you can find 168997at4890_tRNA-splicing_endonuclease_subunit.fasta) and align with high accuracy local alignment setting:
$ singularity exec mafft\:7.525--h031d066_1 mafft --localpair --reorder --maxiterate 1000 --thread 4 data/168997at4890_tRNA-splicing_endonuclease_subunit.fasta > results/168997at4890_tRNA-splicing_endonuclease_subunit_mafft_aligned.fasta
Build the profile
$ singularity pull https://depot.galaxyproject.org/singularity/hmmer:3.4--h503566f_3
$ singularity exec hmmer:3.4--h503566f_3 hmmbuild --cpu 4 --amino ./results/168997at4890_tRNA-splicing_endonuclease_subunit_mafft_aligned.hmm ./results/168997at4890_tRNA-splicing_endonuclease_subunit_mafft_aligned.fasta
TASK 5
Take a look at the profile we just built: Knowing how is the profile file format structured, can you guess what is the most likely amino acid at position 1? Does it make sense?
Search for homologous sequences in the proteome using the profile
It comes from the NCBI annotation of the genome, and is already downloaded in data/. We will HMMER on the proteome (.fasta file with amino-acidic sequences) because hmm profiles only work within the same alphabet (no method like tblastn) -> if you want to search a nucleotide sequence (e.g. assemblies, transcriptome) you need a nucleotide hmm profile
$ singularity exec hmmer\:3.4--h503566f_3 hmmsearch --cpu 4 -E 1e-5 -o ./results/hmmerout-168997_vs_Tmel.txt -A ./results/hmmerout_alignment-168997_vs_Tmel.txt --tblout ./results/hmmerout_table-168997_vs_Tmel.txt ./results/168997at4890_tRNA-splicing_endonuclease_subunit_mafft_aligned.hmm ./data/GCA_000151645.1_ASM15164v1_Tmel_protein.faa
# hmmer output
$ less results/hmmerout-168997_vs_Tmel.txt
# hit(s) as Stockholm alignment
$ less results/hmmerout_alignment-168997_vs_Tmel.txt
# parseable table
$ less results/hmmerout_table-168997_vs_Tmel.txt
Try blastp for comparison
$ singularity exec blast\:2.16.0--h66d330f_4 makeblastdb -in ./data/GCA_000151645.1_ASM15164v1_Tmel_protein.faa -dbtype prot -out ./results/GCA_000151645.1_protein_db
$ singularity exec blast\:2.16.0--h66d330f_4 blastp -num_threads 4 -evalue 1e-5 -query ./data/168997at4890_tRNA-splicing_endonuclease_subunit.fasta -db ./results/GCA_000151645.1_protein_db -out ./results/blastout_168997at4890_protein_vs_GCA_000151645.1_protein_db.txt
$ grep -A 2 "significant" results/blastout_168997at4890_protein_vs_GCA_000151645.1_protein_db.txt
In this case the output is equivalent, except HMMER is able to compare the profile containing all the information of a multifasta, outputting only one alignment, with the advantage of also being faster here (one blastp search alone, would be faster, but less sensitive!). On less straightforward, more variable/distantly related sequences, HMMER methods have an advantage, but for both methods the reference query sequences are crucial.
FINAL TASK
BUILD A REPORT CONTAINING EVERY STEP YOU TOOK, REPORTING THE COMMANDS USED AND THEIR OUTPUT (meaningful examples or summary tables are enough if the output is large!). IT CAN BE DELIVERED IN .docx, .pdf, OR MARKDOWN TEXT FILE.
Does the basidiomycetes fungus Tremella fuciformis (aka white jelly mushroom, snow ear or and silver ear fungus wiki page) have the genetic potential of doing meiosis?
Hints:
Look for the so called meiosis genes in eukaryotes. For example in Ramesh et al., 2005 or Schurko & Logsdon, 2008. You will need gene names.
Query (NCBI) for the genes in close organisms (as we did in TASK1). Help for taxonomy of the target organism at NCBI taxonomy (useful for any organism) or https://www.mycobank.org/ (specific for Fungi)
Search for the genome assembly of T. fuciformis (
datasets downloadcommand, or NCBI webpage).Set up suitable BLAST searches using what you learnt so far (nucleotide, protein query)
For multiple repeated commands, launched over many files is convenient to set up a
forloop (look at the many examples along the course) and/or blast using multifasta files as query.Starting from the same gene set you built, build an .hmm profiles and use HMMER3 to search for the same meiosis genes in the genome assembly of T. fuciformis.
Questions (Read them carefully before starting and use them to guide your analyses)
Did you find more hits on the meiosis genes using BLAST with nucleotide or amino acid sequences as query? With what setting?
What should work better for remote homology, Why?
Verify in the blast output if meiosis genes are in single copy. Is any of your blast search finding hits in multiple positions in the genome assembly? Are they located on the same chromosome? Is the genome assembly of T. fuciformis good enough to verify this?
What are the advantages/disadvantages of HMMER in comparison to BLAST? Could you use HMMER with protein profiles in this case?
What analysis would need to be performed on the genome assembly before using protein hmm profile in search of possible homologs of these meiosis genes?
Compare the the two approaches (in term of method method, number of genes obtained, length of the hits).
What you found looking at the genomic sequence makes sense biologically? (hint: do you know the biology/ecology of the organism? Gather some information about the target organisms, from both generalist and specialized literature, such as scientific papers searched via Google Scholar or similar search engine).